A DFTB Study on Au36, 37 & 39, 40 (C1) Metallic Gold Nanoclusters (AuNCs), the Double and the Triple-State Degrees of Degeneracy
Received: 16-Dec-2022, Manuscript No. Puljnn-22-5915; Editor assigned: 21-Dec-2022, Pre QC No. Puljnn-22-5915 (PQ); Accepted Date: Mar 10, 2023; Reviewed: 17-Jan-2023 QC No. Puljnn-22-5915 (Q); Revised: 21-Feb-2023, Manuscript No. Puljnn-22-5915 (R); Published: 30-Mar-2023, DOI: 10.37532/puljnn.2023.7(1).1-5.
Citation: K Vishwanathan. A DFTB study on Au36, Au37, Au39, and Au40 (C1) metallic gold nanoclusters (AuNCS), the double and the triple-state degrees of degeneracy. J Nanosci Nanomed 2023; 7(1): 01-05
This open-access article is distributed under the terms of the Creative Commons Attribution Non-Commercial License (CC BY-NC) (http://creativecommons.org/licenses/by-nc/4.0/), which permits reuse, distribution and reproduction of the article, provided that the original work is properly cited and the reuse is restricted to noncommercial purposes. For commercial reuse, contact reprints@pulsus.com
Abstract
In this article, an interesting phenomenon has described the geometries and vibrational frequency of the stable AuN clusters with N=36, 37, 39 and 40. We have found all 4 clusters are having the very same C1 point symmetry group. For the re-optimization process, the finite-differentiation method has been implemented within the Density Functional Tight Binding (DFTB) approach. The effects of the range of interatomic forces were calculated and the desired set of system Eigen frequencies (3N-6) are obtained by diagonalization of the symmetric positive semidefinite Hessian matrix. More than anything else, we have observed the vibrational spectra, which occur between 0.98 cm-1 and 304.48 cm-1 at ∆E =0. Most significantly, all the clusters had come across the double and the triple state degeneracies, which are due to the stretching and the bending mode of the vibrations through the atoms. Nevertheless, the vibrational spectrum is strongly dependent upon size, shape, and structure.
Key Words
Gold atomic clusters; Density-functional tight-binding; Finite-difference method; Force constants; Vibrational spectrum
Introduction
Nanoclusters have potential uses in chemical reactors, telecommunications, microelectronics, optical data storage, catalysts magnetic storage, spintronic devices, electroluminescent displays, sensors, biological markers, switches, nano-electronics, nano-optics, transducers and many other fields [1, 2]. Noble metal like Rhodium (Rh), Palladium (Pd), silver (Ag), Platinum (Pt), and Gold (Au) is one kind of modish and desired material, according to their inherent resistance to oxidation and corrosion even in the moist environment [3-5]. Its physical and chemical properties appear to be entirely change as the size of metal continuously decreases into nanoscale because of the quantum size effect, surface effect, small size effect, and Macroscopic Quantum Tunnelling (MQT) effect [6-8].
In general, Noble metal (Cu, Ag, and Au) clusters have attracted much attention in scientific and technological fields because of their thermodynamic, electronic, optical and catalytic properties in nano-materials [9-11]. Especially, gold is a soft metal and is usually alloyed to give it more strength as well as a good conductor of heat and electricity, and is unaffected by air and most reagents, those are the main reasons to choose among the other metal clusters.
In this study, mainly we focus on the vibrational properties of gold atomic clusters with sizes Au36 , Au37 , Au39 and Au40 atoms, because, the vibrational properties play a major role in structural stability [12-18]. For further assistance for the readers, specifically for the general information about global minima gold structures which have been calculated by the work of Dong and Springborg can be found in those articles [19, 20]. In very short, the structures were found through a so called Genetic Algorithm (GA) in combination with Density Functional Tight Binding (DFTB) energy calculations and the steepest descent algorithm permitting a local total energy minimization. Nevertheless, in our case, we use the numerical finitedifference method along with the Density Functional Tight Binding (DFTB) approach and finally extract the vibrational spectrum from the optimized structures [21]. Overall, for a better understanding and to visualize, the detailed information is discussed in the results and discussion section.
Material and Methods
At first step, the DFTB is based on the density functional theory of Hohenberg and Kohn in the formulation of Kohn and Sham [22-24]. In addition, the Kohn-Sham orbitals ψ i (r) of the system of interest are expanded in terms of atom-centered basis functions {ϕ m(r)},

While so far the variational parameters have been the real-space grid representations of the pseudo wave functions, it will now be the set of coefficients cim. Index m describes the atom, where φmis centered and it is angular as well as radially dependent. The φm is determined by self-consistent DFT calculations on isolated atoms using large Slater-type basis sets.
In calculating the orbital energies, we need the Hamilton matrix elements and the overlap matrix elements. The above formula gives the secular equations

Here, cim’s are expansion coefficients, i ∈ is for the single-particle energies (or where i ∈ are the Kohn-Sham eigenvalues of the neutral), and the matrix elements of Hamiltonian Hmn and the overlap matrix elements Smn are defined as

They depend on the atomic positions and on a well-guessed density ρ (r) . By solving the Kohn-Sham equations in an effective one particle potential, the Hamiltonian ˆH is defined as

To calculate the Hamiltonian matrix, the effective potential Veff has to be
approximated. Here, (ωi) being the kinetic-energy operator 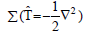 and
Veff(r) being the effective Kohn-Sham potential, which is approximated as a
simple superposition of the potentials of the neutral atoms,
and
Veff(r) being the effective Kohn-Sham potential, which is approximated as a
simple superposition of the potentials of the neutral atoms,

Voj is the Kohn-Sham potential of a neutral atom, rj = r − Rj is an atomic position, and Rj being the coordinates of the jth atom. Finally, the short-range interactions can be approximated by simple pair potentials, and the total energy of the compound of interest relative to that of the isolated atoms is then written as,
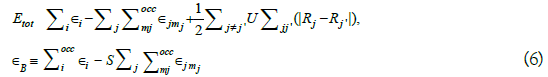
Here, the majority of the binding energy (∈i) is contained in the difference
between the single-particle energies ∈i of the system of interest and the
single-particle energies of the isolated atoms (atom index j, orbital index mj),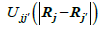 is determined as the difference between ∈B and for diatomic
molecules (with ESCF being the total energy from parameter free density
functional calculations). In the present study, only the 5d and 6s electrons of
the gold atoms are explicitly included, whereas the rest are treated within a
frozen-core approximation [22, 24, 25].
is determined as the difference between ∈B and for diatomic
molecules (with ESCF being the total energy from parameter free density
functional calculations). In the present study, only the 5d and 6s electrons of
the gold atoms are explicitly included, whereas the rest are treated within a
frozen-core approximation [22, 24, 25].
Structural re-optimization process
In our case, we have calculated the numerical first-order derivatives of the forces (Fiα, Fjβ) instead of the numerical-second-order derivatives of the total energy (Etot). In principle, there is no difference, but numerically the approach of using the forces is more accurate [21].
Here, F is a restoring forces which is acting upon the atoms, ds is a differentiation step-size and M represents the atomic mass, for homonuclear case. The complete list of these force constants (FCs) is called the Hessian H, which is a (3N×3N) matrix. Here, i is the component of (x, y or z) of the force on the jth atom, so we get 3N [26].
Results and Discussion
The optimized structure of the clusters Au36, Au37, Au39 and Au40
We present the vibrational spectrum analysis of the re-optimized Au36, Au37, Au39 and Au40 clusters, interestingly, all of them are having the very same point group symmetry C1 at ground state, ΔE= 0. Initially, the structures were found through a so-called Genetic Algorithm (GA) in combination with Density Functional Tight-Binding (DFTB) energy calculations and the steepest descent algorithm permitting a local total energy minimization [19]. To sum up, we have accurately predicted the vibrational frequency of the clusters, and they are very strongly dependent on the size, structure, and shape of the clusters, mainly influenced by the stretching and the bending mode vibrations of the atoms that are due to changes on the bond length fluctuations for a small step-size ds= ± 0.01 a.u. on the equilibrium coordinates [27]. By the way, for the perspective view of the structures, we have plotted with two different styles (Space-filling, Polyhedral).
The vibrational frequency (ωi) range of the cluster Au36 at ΔE = 0
Table 1 shows the low (at the least) and the high (at the most) frequency range of the cluster Au36, which occurs between 4.38 cm-1 and 302.78 cm-1, and the lowest energy geometrical structural view can be seen in Figure 1. Firstly, the cluster has some low frequencies (ωmin) in between 4.38 cm-1 to 8.34 cm-1, which is only for the very first 4 NVM that comes even below the scale of Far Infrared (FIR), IR-C 200 cm-1 to 10 cm-1. Secondly, for the 5 NVM to 84 NVM, the frequency ranges occurred between 10.02-198.10 cm-1, which comes within the range of Far Infrared FIR, IR-C 200cm-1 to 10 cm-1. Thirdly, the rest of the 85 NVM to 102 NVM, is having the maximum high frequencies, which are ((ωi) 200.71 cm-1 to 302.78 cm-1) falling within the range of Mid Infrared MIR, IR-C 3330 cm-1 to 200 cm-1.
Table 1. The Normal modes (NVM) and the vibrational frequencies (ωi) of Au36 at ∆E=0
| NVM (3N-6) | ωi [cm−1] | NVM (3N-6) | ωi[cm−1] | NVM (3N-6) | ωi [cm−1] | NVM (3N-6) | ωi [cm−1] |
|---|---|---|---|---|---|---|---|
| 1 | 4.38 | 30 | 44.97 | 59 | 108.3 | 88 | 212.4 |
| 2 | 5.97 | 31 | 46.02 | 60 | 112.58 | 89 | 214.94 |
| 3 | 7.62 | 32 | 47.1 | 61 | 116.64 | 90 | 221.29 |
| 4 | 8.34 | 33 | 47.67 | 62 | 119.95 | 91 | 221.84 |
| 5 | 10.02 | 34 | 49.62 | 63 | 121.54 | 92 | 226.14 |
| 6 | 10.38 | 35 | 51.6 | 64 | 125.09 | 93 | 232.36 |
| 7 | 12.94 | 36 | 52.79 | 65 | 130.95 | 94 | 235.23 |
| 8 | 13.11 | 37 | 54.81 | 66 | 135.05 | 95 | 239.78 |
| 9 | 14.39 | 38 | 57.29 | 67 | 137.05 | 96 | 245.21 |
| 10 | 15.62 | 39 | 58.96 | 68 | 141.51 | 97 | 249.46 |
| 11 | 17.16 | 40 | 59.72 | 69 | 143.49 | 98 | 255.07 |
| 12 | 17.85 | 41 | 64.46 | 70 | 147.07 | 99 | 266.58 |
| 13 | 19.29 | 42 | 65.25 | 71 | 151.47 | 100 | 269.13 |
| 14 | 20.49 | 43 | 67.39 | 72 | 155.24 | 101 | 279.52 |
| 15 | 21.76 | 44 | 68.25 | 73 | 159.17 | 102 | 302.78 |
| 16 | 23.96 | 45 | 70.27 | 74 | 165.82 | 103 | - |
| 17 | 25 | 46 | 74.38 | 75 | 168.54 | 104 | - |
| 18 | 25.64 | 47 | 76.33 | 76 | 169.41 | 105 | - |
| 19 | 27.39 | 48 | 78.39 | 77 | 177.04 | 106 | - |
| 20 | 28.49 | 49 | 79.86 | 78 | 177.85 | 107 | - |
| 21 | 29 | 50 | 81.29 | 79 | 181.16 | 108 | - |
| 22 | 30.13 | 51 | 82.99 | 80 | 182.49 | 109 | - |
| 23 | 32.57 | 52 | 86.57 | 81 | 186.85 | 110 | - |
| 24 | 33.18 | 53 | 88.4 | 82 | 194.38 | 111 | - |
| 25 | 34.85 | 54 | 92.48 | 83 | 196.94 | 112 | - |
| 26 | 35.92 | 55 | 95.09 | 84 | 198.1 | 113 | - |
| 27 | 37.48 | 56 | 98.51 | 85 | 200.71 | 114 | - |
| 28 | 39.36 | 57 | 101.53 | 86 | 203.73 | 115 | - |
| 29 | 43.59 | 58 | 103.79 | 87 | 209.54 | 116 | - |

Figure 1: Au36 (C1); Style (Space-filling [up], Polyhedral [down]): The lowest energy geometrical structure of the Au36 cluster. Standard orientation of crystal shape at ΔE=0.
The double state degeneracy (ωi)
[{10.02, 10.38}; {17.16, 17.85}; {25.00, 25.64}; {47.10, 47.67}; {177.04, 177.85} and {221.29, 221.84}] in cm-1
The triple state degeneracy (ωi)
We do not find anything; it is clearly revealed through our spectrum calculations that have not occurred.
The vibrational frequency (ωi) range of the cluster Au37 at ΔE=0
Table 2 shows the low (at the least) and the high (at the most) frequency range of the cluster Au37, which occurs between 4.70 cm-1 and 300.67 cm-1, and the lowest energy geometrical structural view can be seen in Figure 2. Firstly, the cluster has some low frequencies (ωmin) between 4.70 cm-1 to 9.59 cm-1, which is only for the very first 6 NVM that comes even below the scale of Far Infrared FIR, IR-C 200 cm-1 to 10 cm-1. Secondly, for the 7 NVM to 87 NVM, the frequency ranges occurred between 10.44 cm-1 to 197.79 cm-1, which comes within the range of Far Infrared FIR, IR-C 200 cm-1 to 10 cm-1. Thirdly, the rest of the 88 NVM to 105 NVM, is having the maximum high frequencies, which are ((ωi) 201.54 cm-1 to 300.67 cm-1) falling within the range of Mid Infrared MIR, IR-C 3330 cm-1 to 200 cm-1.
TABLE 2 The Normal modes (NVM) and the vibrational frequencies (ωi) of Au37 at ∆E=0
| NVM | ωi | NVM | ωi | NVM | ωi | NVM | ωi |
|---|---|---|---|---|---|---|---|
| (3N-6) | [cm−1] | (3N-6) | [cm−1] | (3N-6) | [cm−1] | (3N-6) | [cm−1] |
| 1 | 4.7 | 30 | 40.44 | 59 | 95.98 | 88 | 201.54 |
| 2 | 5.57 | 31 | 41.58 | 60 | 100.6 | 89 | 203.22 |
| 3 | 6.12 | 32 | 43.18 | 61 | 103.21 | 90 | 206.19 |
| 4 | 6.48 | 33 | 43.59 | 62 | 106.95 | 91 | 210.95 |
| 5 | 7.25 | 34 | 44.78 | 63 | 107.99 | 92 | 214.22 |
| 6 | 9.59 | 35 | 46.75 | 64 | 111.04 | 93 | 216.98 |
| 7 | 10.44 | 36 | 48.3 | 65 | 115.47 | 94 | 219.55 |
| 8 | 10.62 | 37 | 49.48 | 66 | 118.7 | 95 | 223.47 |
| 9 | 11.72 | 38 | 51.14 | 67 | 120.12 | 96 | 228.96 |
| 10 | 13.03 | 39 | 52.11 | 68 | 123.96 | 97 | 236.19 |
| 11 | 14.85 | 40 | 54.23 | 69 | 127.65 | 98 | 244.5 |
| 12 | 16.74 | 41 | 56.77 | 70 | 130.9 | 99 | 257.14 |
| 13 | 17.18 | 42 | 57.68 | 71 | 139.09 | 100 | 264.55 |
| 14 | 17.87 | 43 | 59.16 | 72 | 142.88 | 101 | 267.19 |
| 15 | 19.16 | 44 | 62.37 | 73 | 146.46 | 102 | 274.03 |
| 16 | 22.28 | 45 | 63.86 | 74 | 154.56 | 103 | 286.27 |
| 17 | 23.29 | 46 | 66.45 | 75 | 156.19 | 104 | 294.69 |
| 18 | 24.98 | 47 | 69 | 76 | 159.96 | 105 | 300.67 |
| 19 | 26.61 | 48 | 70.08 | 77 | 163.89 | 106 | - |
| 20 | 28.5 | 49 | 72.15 | 78 | 165.34 | 107 | - |
| 21 | 29.07 | 50 | 74.4 | 79 | 167.89 | 108 | - |
| 22 | 29.95 | 51 | 78.31 | 80 | 172.55 | 109 | - |
| 23 | 31.5 | 52 | 80.63 | 81 | 175.83 | 110 | - |
| 24 | 32.12 | 53 | 82.92 | 82 | 185.66 | 111 | - |
| 25 | 33.75 | 54 | 83.89 | 83 | 186.42 | 112 | - |
| 26 | 35.74 | 55 | 86.4 | 84 | 187.35 | 113 | - |
| 27 | 36.46 | 56 | 87.69 | 85 | 189.15 | 114 | - |
| 28 | 37.3 | 57 | 91.87 | 86 | 196.88 | 115 | - |
| 29 | 39.96 | 58 | 95.22 | 87 | 197.79 | 116 | - |

Figure 2: Au37 (C1); Style (Space-filling [left], Polyhedral [right]): The lowest energy geometrical structure of the Au37 cluster. Standard orientation of crystal shape at ΔE=0.
The double and the triple state degeneracy (ωi)
[{6.12, 6.48}; {10.44, 10.62}; {17.18, 17.87}; {29.07, 29.95}; {43.18, 43.59} and {95.22, 95.98}] in cm-1.
The triple state degeneracy (ωi)
We do not find anything; it is clearly revealed through our spectrum calculations that have not occurred.
The vibrational frequency (ωi)
range of the cluster Au39 at ΔE=0
Table 3 shows the low (at the least) and the high (at the most) frequency range of the cluster Au39, which occurs between 0.98 and 304.48 cm-1, and the lowest energy geometrical structural view can be seen in Figure 3. Firstly, the cluster has some low frequencies (ωmin) between 0.98 cm-1-9.50 cm-1, which is only for the very first 7 NVM, which comes even below the scale of Far Infrared FIR, IR-C 200 cm-1 to 10 cm-1. Secondly, for the 8 NVM to 94 NVM, the frequency ranges are occurred between 10.38-198.61 cm-1, which comes within the range of Far Infrared FIR, IR-C 200- 10 cm-1. Thirdly, for the rest of the 95 NVM to 111 NVM, are having the maximum high frequencies, which are ((ωi)-202.26 cm-1 to 304.48 cm-1) falling within the range of Mid Infrared MIR, IR-C 3330 cm-1 to 200 cm-1.
TABLE 3 The Normal Modes (NVM) and the vibrational frequencies (ωi) of Au39 at ΔE=0
| NVM | ωi | NVM | ωi | NVM | ωi | NVM | ωi |
|---|---|---|---|---|---|---|---|
| (3N-6) | [cm−1] | (3N-6) | [cm−1] | (3N-6) | [cm−1] | (3N-6) | [cm−1] |
| 1 | 0.98 | 30 | 35.47 | 59 | 88.84 | 88 | 178.33 |
| 2 | 2.45 | 31 | 36.2 | 60 | 90.61 | 89 | 184.04 |
| 3 | 3.65 | 32 | 38.72 | 61 | 93.88 | 90 | 188.67 |
| 4 | 6.26 | 33 | 39.71 | 62 | 94.48 | 91 | 189.62 |
| 5 | 7.3 | 34 | 41.64 | 63 | 98.71 | 92 | 191.1 |
| 6 | 8.81 | 35 | 42.88 | 64 | 101.56 | 93 | 195.07 |
| 7 | 9.5 | 36 | 43.47 | 65 | 103.1 | 94 | 198.61 |
| 8 | 10.38 | 37 | 44.93 | 66 | 104.42 | 95 | 202.26 |
| 9 | 11.12 | 38 | 46.37 | 67 | 107.12 | 96 | 206.97 |
| 10 | 12.52 | 39 | 48.47 | 68 | 108.12 | 97 | 209.26 |
| 11 | 12.78 | 40 | 49.33 | 69 | 113.48 | 98 | 212.2 |
| 12 | 13.91 | 41 | 49.77 | 70 | 117.12 | 99 | 218.05 |
| 13 | 14.98 | 42 | 51.37 | 71 | 122.33 | 100 | 222.72 |
| 14 | 16.82 | 43 | 53.25 | 72 | 125.21 | 101 | 225.94 |
| 15 | 18.3 | 44 | 53.67 | 73 | 126.61 | 102 | 226.95 |
| 16 | 18.97 | 45 | 56.07 | 74 | 132.96 | 103 | 229.29 |
| 17 | 20.05 | 46 | 56.83 | 75 | 135.78 | 104 | 232.75 |
| 18 | 20.35 | 47 | 58.43 | 76 | 138.05 | 105 | 241.6 |
| 19 | 22.63 | 48 | 60.97 | 77 | 139.61 | 106 | 248.58 |
| 20 | 22.94 | 49 | 61.77 | 78 | 143.6 | 107 | 254.79 |
| 21 | 23.04 | 50 | 63.74 | 79 | 147.6 | 108 | 259.57 |
| 22 | 25.16 | 51 | 66.17 | 80 | 148.45 | 109 | 269.84 |
| 23 | 25.93 | 52 | 67.94 | 81 | 155.8 | 110 | 279.22 |
| 24 | 27.88 | 53 | 70.88 | 82 | 157.65 | 111 | 304.48 |
| 25 | 28.21 | 54 | 73.05 | 83 | 163.5 | 112 | - |
| 26 | 29.16 | 55 | 76.56 | 84 | 168.95 | 113 | - |
| 27 | 30.2 | 56 | 77.91 | 85 | 172.36 | 114 | - |
| 28 | 32.55 | 57 | 82.53 | 86 | 173.81 | 115 | - |
| 29 | 33.49 | 58 | 83.5 | 87 | 175.25 | 116 | - |

Figure 3: Au39 (C1); Style (Space-filling [left], Polyhedral [right]): The lowest energy geometrical structure of the Au39 cluster. Standard orientation of crystal shape at ΔE=0
The double and the triple state degeneracy (ωi)
[{12.52, 12.78}; {18.30, 18.97}; {20.05, 20.35}; {22.63, 22.94}; {25.16, 25.93}; {49.33, 49.77}; {53.25, 53.67} and {56.07 56.83}] in cm-1.
The triple state degeneracy (ωi)
We do not find anything; it is clearly revealed through our spectrum calculations that have not occurred.
The vibrational frequency (ωi) range of the cluster Au40 at ΔE=0
Table 4 shows the low (at the least) and the high (at the most) frequency range of the cluster Au40, which occurs between 2.55 cm-1 and 282.94 cm-1, and the lowest energy geometrical structural view can be seen in Figure 4. Firstly, the cluster has some low frequencies (ωmin) between 2.55 cm-1 to 9.39 cm-1, which is only for the very first 6 NVM, which comes even below the scale of Far Infrared FIR, IR-C 200 cm-1 to 10 cm-1. Secondly, for the 7NVM to 95 NVM, the frequency ranges are occurred between 10.65 cm-1 to 195.06 cm-1, which comes within the range of Far Infrared FIR, IR-C 200 cm-1 to 10 cm-1. Thirdly, the rest of the 96 NVM to 114 NVM, are having the maximum high frequencies, which are ((ωi)-202.65 cm-1 to 282.94 cm-1) falling within the range of Mid Infrared MIR, IR-C 3330 cm-1 to 200 cm-1.
TABLE 4 The Normal modes (NVM) and the vibrational frequencies (ωi) of Au40 at ΔE=0.
| NVM | ωi | NVM | ωi | NVM | ωi | NVM | ωi |
|---|---|---|---|---|---|---|---|
| (3N-6) | [cm−1] | (3N-6) | [cm−1] | (3N-6) | [cm−1] | (3N-6) | [cm−1] |
| 1 | 2.55 | 30 | 34.85 | 59 | 83.68 | 88 | 174.29 |
| 2 | 4.14 | 31 | 35.97 | 60 | 84.14 | 89 | 179.75 |
| 3 | 6.12 | 32 | 37.67 | 61 | 87.77 | 90 | 180.67 |
| 4 | 7.81 | 33 | 38.89 | 62 | 89.33 | 91 | 187.99 |
| 5 | 8.54 | 34 | 38.94 | 63 | 91.82 | 92 | 189.46 |
| 6 | 9.39 | 35 | 40.42 | 64 | 96.66 | 93 | 192.24 |
| 7 | 10.65 | 36 | 41.04 | 65 | 99.94 | 94 | 192.68 |
| 8 | 11.1 | 37 | 42.64 | 66 | 100.12 | 95 | 195.06 |
| 9 | 11.98 | 38 | 44.83 | 67 | 100.89 | 96 | 202.65 |
| 10 | 13.19 | 39 | 46.41 | 68 | 103.57 | 97 | 206.41 |
| 11 | 13.52 | 40 | 48.61 | 69 | 111.14 | 98 | 210.63 |
| 12 | 15.13 | 41 | 49.33 | 70 | 114.92 | 99 | 214.72 |
| 13 | 17.71 | 42 | 51.04 | 71 | 118.04 | 100 | 221.2 |
| 14 | 18.41 | 43 | 52.27 | 72 | 120.91 | 101 | 223.58 |
| 15 | 18.6 | 44 | 54.05 | 73 | 123.88 | 102 | 228.32 |
| 16 | 20.17 | 45 | 56.5 | 74 | 127.89 | 103 | 230.08 |
| 17 | 20.92 | 46 | 58.92 | 75 | 131.06 | 104 | 231.76 |
| 18 | 23.34 | 47 | 60.24 | 76 | 135.96 | 105 | 238.29 |
| 19 | 23.66 | 48 | 60.67 | 77 | 138.51 | 106 | 239.02 |
| 20 | 24.19 | 49 | 64.16 | 78 | 141.84 | 107 | 244.35 |
| 21 | 25.6 | 50 | 64.34 | 79 | 144.78 | 108 | 246.71 |
| 22 | 26.58 | 51 | 68.97 | 80 | 151.84 | 109 | 253.61 |
| 23 | 27.46 | 52 | 69.76 | 81 | 155.03 | 110 | 254.71 |
| 24 | 28.47 | 53 | 70.81 | 82 | 158.68 | 111 | 263.59 |
| 25 | 29.86 | 54 | 72.22 | 83 | 159.92 | 112 | 266.41 |
| 26 | 31.03 | 55 | 73.45 | 84 | 165.21 | 113 | 270.79 |
| 27 | 31.58 | 56 | 76.78 | 85 | 165.61 | 114 | 282.94 |
| 28 | 33.57 | 57 | 79.49 | 86 | 171.11 | 115 | - |
| 29 | 33.68 | 58 | 81.47 | 87 | 172.51 | 116 | - |

Figure 4: Au40 (C1); Style (Space-filling [left], Polyhedral [right]): The lowest energy geometrical structure of the Au40 cluster. Standard orientation of crystal shape at ΔE=0
The double state degeneracy (ωi)
[{11.10, 11.98}; {13.19, 13.52}; {18.41, 18.60}; {20.17, 20.92}; {23.34, 23.66}; {31.03, 31.58}; {33.57, 33.68}; {38.89, 38.94}; {60.24, 60.67}; {64.16, 64.34}; {100.12, 100.89}; {165.21, 165.61} and {192.24, 192.68}] in cm-1.
The triple state degeneracy (ωi)
We do not find anything; it is clearly revealed through our spectrum calculations that have not occurred.
Size and the shape effects
In Table 5, the third column shows the spectral ranges that have been influenced with respect to the size of the clusters, the shape of the structures, and the arrangement of the atoms (inner core, and the overall outer surface of the edges), as well as the short and the long range interactions due to the inter-nuclear attraction and the repulsive energies.
TABLE 5 The double and the triple state degree of degeneracy of the clusters, Au36, Au37, Au39 and Au40 at ΔE = 0. Unfortunately, we do not find anything; it is clearly revealed through our spectrum calculations the triple state degree of degeneracy that has not occurred.
| Gold Nanoclusters (AuNCs) | Point Groups (PG) Symmetry | Spectral Range (min - to -max) ωi [cm-1] | Double (D) & Triple (T) State Degeneracy [DT]{pairs} | Total Number of Pairs | Total Random Number (RN) of Different States of Equal Energy RN=(D*pairs+T*pairs) | Predicted Spectral Range Only for D, T- Degeneracies A: Far Infrared FIR, IR - C 200 - 10 cm-1 B: Mid Infrared MIR, IR - C 4000 - 200 cm-1 X: Lesser than both, A and B |
|---|---|---|---|---|---|---|
| Au36 | C1 | 4.38-302.78 | D6 T0 | 6 | 12 | A, B |
| Au37 | C1 | 4.70-300.67 | D6 T0 | 6 | 12 | A, X |
| Au39 | C1 | 0.98-304.48 | D8 T0 | 8 | 16 | A |
| Au40 | C1 | 2.55-282.94 | D13 T0 | 13 | 26 | A |
Conclusion
We are first to present, the vibrational frequencies of bigger-sized clusters (Au36, Au37, Au39 and Au40) and the shell-like structure of the re-optimized structures, at ΔE=0, by using the numerical finite-differentiation method with the DFTB approach. We found the vibrational spectrum, the minimum starting, and the maximal end ranges that vary between 0.98 cm-1and 304.48 cm-1 at ΔE=0. The occupancy of the multiple double and the triple state degeneracy is revealed on the gold atomic clusters, Au36, Au37, Au39 and Au40. More the double-state degeneracy may depend on the nearest neighboring atoms, and their interactions, as well as the zig-zag circumstances of the outermost surface surrounded by them. We are able to see, a maximum, of 13 total pairs have occurred on the Au40 cluster, which is very special to the others.
Above all, we have pinpointed the correct location of the spectrum, through Far Infrared FIR, IR-C 200 cm-1 to 10 cm-1, and Mid Infrared MIR, IR-C 3330 cm-1 to 200 cm-1, and even outside of this range. In addition to that, our prediction will help the researchers to develop a range of potential applications such as catalysis, biomedicine, imaging, optics, and energy conversion.
References
- Wu L, Fang W, Chen X. The photoluminescence mechanism of ultra-small gold clusters. Phys Chem Chem Phys. 2016;18(26):17320-5.
- Andres RP, Bein T, Dorogi M, et al. Coulomb staircase at room temperature in a self-assembled molecular nanostructure. science. 1996;272(5266):1323-5.
- Griffith WP. The Chemistry of the Rarer Platinum Metals. Inter-Science: New York. 1967:2-4.
- Hartley FR. The chemistry of platinum and palladium: with particular reference to complexes of the elements. Appl Sci Publ; 1973.
- Huang X. Polymer Ligand Stabilized Fluorescent Platinum Nanoclusters: Synthesis, Characterization, and Their Applications. Osaka University. 2016.
- Siegel RW. Nanostructured materials mind over matter. Nanostructured Materials. 1994;4:121-38
- Huang X, Li Z, Yu Z, et al. Recent Advances in the Synthesis, Properties, and Biological Applications of Platinum Nanoclusters. J Nanomater. 2019:6248725.
- Choi YC, Lee HM, Kim WY, et al. How can we make stable linear monoatomic chains? Gold-cesium binary subnanowires as an example of a charge-transfer-driven approach to alloying. Phys Rev lett. 2007;98(7):076101.
- Li J, Liu Y, Zhang J, et al. Density functional theory study of the adsorption of hydrogen atoms on Cu2X (X=3d) clusters. Chem Phys Lett. 2016;651:137-43.
- Chuanchuan Z, Haiming D, Xin L, et al. Staticand dynamical isomerization of Cu38 cluster. Scientific Reports 2019;9:7564.
- Garzon IL, Posada-Amarillas A. Structural and vibrational analysis of amorphous Au 55 clusters. Physical Review B. 1996;15;54(16):11796.
- Bravo-Pérez G, Garzón IL. Novaro O. Ab initio study of small gold clusters. J Mol Struct: THEOCHEM. 1999;15;493(1-3):225-31.
- Bravo-Perez G, Garzon IL, Novaro O. Non-additive effects in small gold clusters. Chem Phys Lett. 1999;313:655-64.
- Sauceda HE, Mongin D, Maioli P, et al. Vibrational properties of metal nanoparticles: Atomistic simulation and comparison with time-resolved investigation. J Phys Chem C. 2012;116:25147-56.
- Sauceda HE, Pelayo JJ, Salazar F, et al. Vibrational spectrum, caloric curve, low-temperature heat capacity, and Debye temperature of sodium clusters: The Na139+case. J Phys Chem C. 2013;117:11393-8.
- Sauceda HE, Salazar F, Perez LA, et al. Size and shape dependence of the vibrational spectrum and low-temperature specific heat of Au nanoparticles. J Phys Chem C. 2013;117:25160-8.
- Sauceda HE, Garzon IL. Structural determination of metal nanoparticles from their vibrational (phonon) density of states. J Phys Chem C. 2015;119:10876.
- Dugan N, Erkoc S. Stability analysis of graphene nanoribbons by molecular dynamics simulations. physica status solidi (b). 2008;245(4):695-700.
- Dong Y, Springborg M. Global structure optimization study on Au2-20. Eur Phys J D. 2007;43:15-18.
- Warnke I. Heat Capacities of Metal Clusters. Diploma Thesis (Research Assistant and Diploma Research), Saarland University. 2007.
- Dvornikov M. Formulae of numerical differentiation. 2004.
- Porezag D, Frauenheim Th, Kohler Th, et al. Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon. Phys Rev B. 1995;51:12947.
- Seifert G, Schmidt R. Molecular dynamics and trajectory calculations: The application of an LCAO-LDA scheme for simulations of cluster-cluster col-lisions. New J Chem. 1992;16:1145.
- Seifert G, Porezag D, Th. Frauenheim. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme. Int J Quantum Chem. 1996;58:185-9.
- Seifert G. Tight-Binding Density Functional Theory: An Approximate Kohn-Sham DFT Scheme. J Phys Chem A. 2007;111:5609-5613.
- Press WH, Teukolsky SA, Vetterling WT, et al. Numerical Recipes in Fortran. Cambridge University Press. 2007
[Google Scholar] [Crossref]
- Vishwanathan K. Bonding Forces and Energies on the Potential Energy Surface (PES) of the Optimized Gold Atomic Clusters by a Differentiation Step-Size (ds = ± 0.01 a.u.) via DFTB Method. Nanosci Technol. 2018;5:1-4.