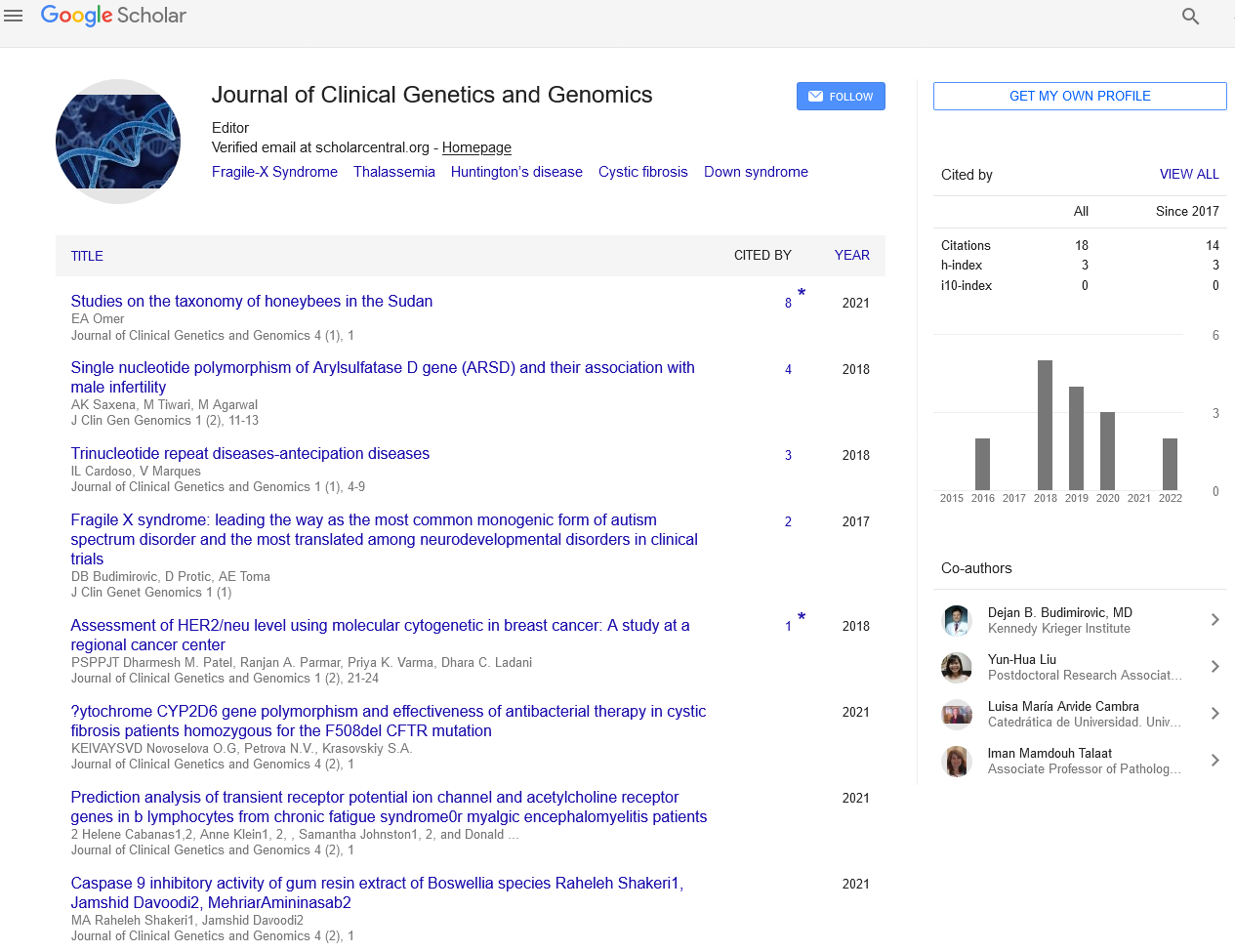
The never ending mystery of genotype-phenotype expression
Received: 14-Nov-2017 Accepted Date: Nov 16, 2017; Published: 27-Nov-2017
Citation: Niyazov DM. The never ending mystery of genotype-phenotype expression. J clin gen genomics. December-2017;1(1):3.
This open-access article is distributed under the terms of the Creative Commons Attribution Non-Commercial License (CC BY-NC) (http://creativecommons.org/licenses/by-nc/4.0/), which permits reuse, distribution and reproduction of the article, provided that the original work is properly cited and the reuse is restricted to noncommercial purposes. For commercial reuse, contact reprints@pulsus.com
We often wonder why it is so difficult to predict a phenotype from one or more reported gene variants, even if their pathogenicity is established. It’s certainly less challenging to sequence DNA than actually clinically interpret what we’ve just sequenced. We may get a variant of unknown significance (VUS) which may segregate with affected family members but whether it’s causative or not would still be uncertain. When we see a “pathogenic variant” (PV) or “likely pathogenic variant” (LPV), we feel that we are very close to or reached the answer. Yet expressivity can be extremely variable and/or penetrance reduced so it’s baffling how the same PV or LPV can hardly touch one family member while another one has a severe disease. We all routinely face the question “what’s going to happen to my child” and are forced to answer that we don’t know for sure especially when a parent has the same variant and virtually no clinical manifestations. It can even happen in identical twins who can be discordant in many different ways.
Neurofibromatosis type 1 (NF1) is a prime example. One of my pediatric patients had cerebellar glioma and body carpeted with café-au-lait spots. His “healthy” father had Lisch nodules thought to be “a cool brown pigment in his blue eyes.” He also had barely visible axillary freckles which everyone thought were the same as myriads of freckles in his face and arms. Both father and son had the same PV in the NF1 gene implicated in the disease considered to have high penetrance. One had no idea he had the disease while the other one was really sick with poor prognosis. When the father was told that it was not his fault, he felt guilty that he escaped with a scratch while his son was struck so badly. He said “I should’ve taken the full brunt, not my son – he’s just a child.” How can we, geneticists, answer this in 2017?
The question of why “same” disease affects two people differently is integral to our understanding of genetics but unfortunately our ability to answer it is woefully inadequate. We base it in part on scientific evidence and on conjectures yet to be proven. To make it more understandable to my patients I make an analogy between the NF1 gene with its thousands of basepairs and a wall with thousands of bricks. The father and son have the same faulty brick while the rest of the bricks are not necessarily the same. They look similar at a distance, but more closely they can have slightly different textures and/or colors. One can be slightly turned while the other may have a thicker seal around it. Some bricks can be marked to be picked for future action while the others may be concealed temporarily. These seemingly insignificant differences make an impact on their function. Perhaps the father’s wall is a bit “sturdier” despite the faulty brick allowing for better functioning under various environmental conditions.
But why in the world would the father’s and son’s genes have base-pairs with differences, my patients ask? After all, isn’t the father supposed to pass the same gene to his son? We now know with certainty that the bricks themselves are passed on the same. But the coloring and flavoring comes from different environmental pressures and gene interactions that are unique starting at conception. As soon as the father’s NF1 gene in the sperm becomes the NF1 gene in the zygote, the base pairs instantaneously begin to acquire different characteristics via the outside changes which are called epigenetic or above genetic.
Going further, it’s not just one brick wall but millions of them that are packed densely into one small space of a cellular nucleus where a DNA molecule made of 3 bln basepairs resides. They are far from being the exact replicas between the father and son. They can have different properties prediposing and/or causing different bricks to become faulty. Some of them confer resistance to tumor formation in the father more so than the son. And then there’re millions more of the bricks (noncoding DNA) and we know even less about them.
An infinite number of potential interactions between genome, epigenome, phenome and environment could only be expressed in an exponential form or essentially infinity. It’s no wonder why we can never entirely predict a phenotype from a genotype even in identical twins with the same primary DNA sequence who are subject to numerous environmental effects altering their DNA differentially starting at conception.
As frustrating as our lack of knowledge sounds to us and our patients, it’s important to emphasize that we have been unraveling these complex interactions between genes and environment particularly since the Human Genome Project. With the massive power of sequencing technology and computing, coupled with scientific research and development, we are getting better at predicting a phenotype from a genotype with increasing clinical relevance to human health. I can never tell my patients that they “will be” or “will have” something by just looking at a lab report. I have to use “may be” or “may have” since false predictions sadly are too common in the world where sequencing outpaced our understanding of what sequencing cannot explain. We have to find understandable ways to educate our patients and colleagues on how immensely complex the interplay of genetics and environment can be and we are getting answers slowly but surely. Even though finding PV or LPV can be a powerful tool, it is only one of many.